
Liposarkom - Liposarcoma
| Liposarkom | |
|---|---|
 | |
| Liposarkom histopatolojisi, H&E boyası: - | |
| uzmanlık | Dermatoloji , genel cerrahi onkoloji |
| Belirtiler | Deri altında yumru, ağrı, şişlik, organ disfonksiyonu |
Liposarkomlar , yetişkinlerdeki tüm sarkomların en az %20'sini oluşturan yumuşak doku sarkomlarının en yaygın alt tipidir . Yumuşak doku sarkomları, 150'den fazla farklı histolojik alt tipi veya formu olan nadir neoplazmlardır . Tüm, yetişkin habis tümörlerinin yaklaşık% 1 oluşturan Sarkomlar, habis gelişen tümörlerin kök hücrelerin bir mezenşimal : (yani, bağ) gibi dokular osteosarkom ortaya osteoprogenitör hücrelerinin (yani, ön-maddesi) olgun hücreleri osteositler kemikte Dokular; fibrosarkom prekürsör hücrelerden ortaya fibrositlerinin olarak bağ dokuları ; ve rabdomiyosarkomlar prekürsör hücrelerden ortaya miyositlerde olarak kas dokuları . Liposarkomlar ön-ortaya çıkan lipoblasts ait adipositler (yani yağ hücreleri) adipoz (örneğin katı yağ) dokuları . Adipoz dokular, deri altı dokuların derin ve daha yüzeysel katmanları gibi bölgelerin yanı sıra retroperiton (yani karın boşluğunun arkasındaki boşluk ) ve karın boşluğu içindeki viseral yağ gibi cerrahi olarak daha az erişilebilen bölgeler de dahil olmak üzere vücudun her yerine dağılır .
Tüm liposarkomlar, mikroskop altında histopatolojik görünümleri açısından incelendiğinde yağ hücrelerine benzerlik gösteren en azından bazı hücrelerden oluşur . Bununla birlikte, liposarkomların klinik görünümlerindeki farklılıklara (örn. yaşlar, cinsiyet tercihleri, tümör bölgeleri, belirtiler ve semptomlar ), ciddiyetlerine (örn. lokal dokuları invaze etme , cerrahi olarak çıkarıldıktan sonra tekrarlama ve distale metastaz yapma potansiyeli) göre çeşitli formları vardır. dokular), genetik anormallikler , prognozlar ve tercih edilen tedavi rejimleri. Dünya Sağlık Örgütü 2020 yılında, beş veya daha az belirgin formlara liposarkomlar sınıflandırılmıştır: 1) lipomatöz tümör / iyi derecede diferansiye liposarkom atipik; 2) farklılaşmamış liposarkom; 3) miksoid liposarkom; 4) pleomorfik liposarkom; ve 5) miksoid pleomorfik liposarkom. ( Pleomorfik , boyut ve şekillerinde ve/veya çekirdeklerinin boyut ve şeklinde anormal ve genellikle büyük varyasyonlara sahip hücrelerin varlığını gösterir.)
Liposarkom formları agresif ve malign olarak sınıflandırılırken veya atipik lipomatöz tümör/iyi diferansiye liposarkom durumunda nispeten agresif olmayan ve iyi huylu olarak sınıflandırılırken , beş liposarkom formunun tamamı yakındaki dokulara ve organlara zarar vermek için lokal olarak infiltre olabilir. hayati organlara bitişik cerrahi olarak erişilemeyen bölgeler (örneğin retroperiton), cerrahi olarak çıkarıldıktan sonra tekrarlar ve yaşamı tehdit eden hastalıklara ilerler. Bugüne kadar yapılan çalışmalar, beş liposarkom formunun tümünün, genellikle en azından başlangıçta cerrahi rezeksiyonla tedavi edilebilmesine rağmen, şu anda kullanılan kemoterapi ve radyoterapi rejimlerine genellikle yalnızca marjinal yanıt verdiğini bulmuştur. Liposarkomlar, çeşitli radyoterapi , kemoterapi ve daha yeni tedavi rejimlerine bireysel olarak ve mümkünse cerrahi olarak çıkarmayı da içeren çeşitli kombinasyonlarda yanıt vermelerini belirlemek için geniş bir yelpazede ileri çalışmalara ihtiyaç duyar .
Liposarkom formları
Liposarkomlar genellikle büyük tümörlerdir (>10 cm), ancak hemen hemen her boyutta olabilirler. Çoğunlukla yetişkinlerde görülür ve vakaların sadece %0.7'si <16 yaşındadır. Yetişkinlerde, liposarkomlar ağırlıklı olarak orta yaşta ve sonrasında ortaya çıkar. Çocuklarda ve ergenlerde ortaya çıkan çok nadir vakalar, ağırlıklı olarak miksoid liposarkom formu olarak teşhis edilir.
Beş liposarkom formu sadece birbirinden değil, aynı zamanda diğer bazı yumuşak doku tümörlerinden de ayırt edilmelidir. Bu diğer tümörler ve bazı ayırt edici histopatolojik özellikleri şunlardır: 1) displastik lipomlar (yani doku nekrozu bölgelerine sahip iyi huylu hümörler ve neoplastik, değişken boyutlu/şekilli çekirdekler içeren değişken boyutlu yağ hücreleri ; çoğu neoplastik hücrenin aksine bu neoplastik hücreler liposarkomlar olarak, yapma aşırı ifade eden MDM2 ) geni; 2) atipik iğ hücreli lipomlar (yani, fibröz-miksoid stromada vakuollü lipoblastlar ve atipik çekirdekli değişken boyutlu adipositler ile karıştırılmış hafif atipik iğ şekilli hücrelere sahip iyi huylu tümörler ; 3) pleomorfik lipomlar (yani dev hücrelerle karakterize iyi huylu tümörler) örtüşen çekirdekler ile); ve 4) soliter fibröz tümörler (yani, karakteristik bir geyik boynuzu şekline sahip kan damarları ile karıştırılmış bir kolajen arka plan stroması içinde iğsi veya oval şekilli hücrelerden oluşan, %22'ye kadarı kötü huylu davranış sergileyen tümörler).
Atipik lipomatöz tümör/iyi diferansiye liposarkom
Atipik lipomatöz tümörler (ALT'ler) ve iyi farklılaşmış liposarkomlar (WDL'ler) birlikte tüm liposarkomların %40 ila %45'ini oluşturur. Nadiren metastaz yaparlar ve bu nedenle benign veya premalign tümörler olarak kabul edilirler . Bununla birlikte, lokal olarak invazivdirler ve daha agresif ve potansiyel olarak metastaz yapan bir liposarkoma, yani farklılaşmamış bir liposarkoma dönüşebilirler . Ayrıca, cerrahi olarak çıkarılmış bir atipik lipomatöz tümör/iyi farklılaşmış liposarkom, farklılaşmış bir liposarkom olarak tekrarlayabilir.
Sunum

ALT'ler ve WDL'ler, tanım gereği ALT'ler kollarda veya bacaklarda gelişen tümörleri belirtirken , WDL'ler retroperitonun derin, merkezi yerleşimli yumuşak dokuları , paratestiküler bölge gibi cerrahi olarak daha az erişilebilir bölgelerde gelişen tümörleri belirtmesi dışında hemen hemen aynı tümörler olarak kabul edilir ( yani testisler , spermatik kord , testis tuniği , epididim ve testis eki dahil skrotum içindeki alan ), ağız boşluğu ve göz yuvası . Bu terminolojinin prognostik etkileri vardır: ALT tümörlerinin %7'sinden azı medyan 7 yıllık bir süre içinde farklılaşmış liposarkoma dönüşürken, WDL tümörlerinin %17'si medyan 8 yıllık bir süre içinde bu daha malign liposarkoma dönüşür. ALT ve WDL (bundan böyle ALT/WDL olarak anılacaktır) tümörleri tipik olarak orta yaşlı ve yaşlı bireylerde, derin dokularda yerleştiğinde daha büyük ve daha ileri bir aşamada olma eğiliminde olan yavaş büyüyen kitleler olarak bulunur. Bu tümörler genellikle ağrılı değildir ve yüzeysel olarak yerleştirildiyse kolayca görülür; bunlar ayrıca , kana ve/veya tümörün bulunduğu bölgeyi boşaltan lenf damarlarına invaze olmaları nedeniyle uyluk gibi ilgili alanlarda (yandaki şekle bakınız) geniş ödemlere (yani lokal sıvı birikiminden dolayı şişmeye) neden olabilirler . Derin yerleşimli ALT/WDL tümörleri asemptomatik olabilir, ancak yerlerine bağlı olarak infiltre ettikleri çeşitli organlardan herhangi birinde ciddi işlev bozukluğu belirtileri ve/veya semptomları üretirler. Bu organlar arasında retroperitona yakın veya içinde bulunanlar (örneğin, bağırsaklar, böbrek ve böbrek üreterleri ); paratestiküler bölge; mediasten (örneğin trakea ve akciğerin ana bronşları ); ve kafa (örneğin, göz küresinin arkasındaki retrobulbar boşluk).
Patoloji

Histopatolojik olarak, ALT/WDL tümörleri adiposit/lipoma benzeri, sklerozan ve en yaygın olarak adiposit/lipoma benzeri olmak üzere inflamatuar varyantlara ayrılır. Adipositik/lipoma benzeri ALT/WDL tümörleri, düzensiz fibröz septa ile değişken şekilde kesişen olgun yağ hücrelerinin lobüllerinden oluşur ( bitişik H&E lekeli fotomikrografiye bakın ). İkinci en yaygın varyant olan sklerozan ALT/WDL tümörleri öncelikle retroperitoneal ve paratestiküler alanlarda gelişir; kolajenli (yani kolajen içeren) bir stromal doku arka planı içinde dağınık, atipik stromal hücrelerden oluşur . Nadir vakuol içeren lipoblastlar bu dokuyu doldurur. Enflamatuar ALT/WDL tümörleri en nadir görülen varyanttır. en sık olarak retroperitonda meydana gelirler ve örneğin lenfositler ve plazma hücreleri gibi kronik enflamatuar hücrelerden ve ayrıca yağ hücrelerini içeren bir doku arka planı boyunca serpiştirilmiş ara sıra lenf düğümü benzeri foliküllerden oluşurlar .
Genetik
ALT/WDL tümörlerindeki neoplastik hücreler, bir veya daha fazla ekstra halka şeklinde küçük süpernumerary markör kromozomu (sSMC) veya anormal bir dev markör kromozomu (yani, kendisinin veya birinin parçalarının kopyalanmasıyla anormal hale getirilen önceden normal bir kromozom) içerir. veya daha fazla başka kromozomun genetik materyali). Bu anormal kromozom ek kopyalarını içeren kromozom 12 'in, uzun kol (olarak da adlandırılır q kolu de) bantlar ile 15 kromozom 12'nin bu streç 13 içerir MDM2 proto-onkogen (potansiyel tümör neden olan gen aşın bandında bulunan) 15 ve CDK4 (aşırı ifade edildiğinde çeşitli tümörlerin gelişimini destekleyen bir gen) 14.1 bandında yer alır. Amplificiation bu iki genin (örneğin, diğer genlerde bir oranda bir artış olmaksızın, bir genin kopya artış) bir liposarkom ya bir ALT / DSK'nin veya dediferansiye liposarkoma yerine başka bir liposarkom veya bir son derece hassas ve spesifik göstergesidir lipom formu. MDM2 ve CDK4 genlerine ek olarak , bu 13-15 bant kromozom alanı , aşırı eksprese edildiğinde çeşitli tümörler ve/veya kanserlerle ilişkili olan TSPAN31 ve HMGA2 genlerini de içerir . Bu aşırı eksprese edilmiş genlerin bir veya daha fazlasının ALT/WDL tümörlerinin gelişimini ve/veya ilerlemesini önerdiği, desteklediği ve/veya katkıda bulunduğu öne sürülmüştür.
Teşhis
ALT/WDL tümörlerinin tanısı klinik görünüm, histopatoloji ve genetik bulguların özelliklerine göre konur. Özellikle, aşırı eksprese edilmiş bir MDM2 veya CDK4 geninin ALT/WDL tümör hücrelerinde tespiti veya spesifik ALT/WDL ile ilişkili sSMC veya dev markör kromozomunun ( yeni nesil DNA dizilimi , karşılaştırmalı genomik hibridizasyon ve/ veya yüksek düzeyde özelleşmiş sitogenetik G bantlama analizleri ), ALT/WDL veya farklılaşmamış liposarkom tanısını güçlü bir şekilde destekler. Son iki liposarkom formu arasındaki klinik görünüm ve histopatolojik farklılıklar genellikle aralarında ayrım yapılmasına yardımcı olur.
Tedavi ve prognoz
ALT/WDL tümörleri, tüm tümör neoplastik dokularını çıkarmak için radikal cerrahi rezeksiyonla tedavi edilir. Bununla birlikte, bu tümörler vakaların %30-50'sinde lokal olarak nüks eder. Nüksler en sık retroperiton, mediasten ve spermatik kord gibi daha az erişilebilir bölgelerde bulunan tümörlerde görülür. Bu daha az cerrahi olarak değerlendirilebilir tümörler, tekrar tekrar tekrarlama eğilimindedir ve hayati organlar üzerindeki zararlı etkileri nedeniyle nihayetinde ölüme neden olabilir. ALT/WDL tümörlerinin metastaz yapma potansiyeli çok az olsa da , yaklaşık %10'u açıkça malign ve potansiyel olarak metastaz yapan bir liposarkom formuna, farklılaşmamış liposarkoma dönüşecektir. Bu malign dönüşümün medyan süresi yaklaşık 7-9 yıldır. Ek olarak, cerrahi olarak çıkarılan bir ALT/WDL, farklı bir liposarkom olarak değişken bir aralıktan sonra tekrarlayabilir. ALT/WDL tümörlerinde radyoterapiyi takiben cerrahi ile tek başına cerrahiyi karşılaştıran büyük bir randomize kontrollü çalışma , iki rejim arasında çok az fark bulmuştur. ALT/WDL'de yer alan CDK4 veya MDM2 genlerinin protein ürünlerinin seçici inhibitörlerini kullanan daha küçük çalışmalar , en iyi ihtimalle sadece mütevazı etkiler göstermiştir. Bu veya tamamen yeni tedavi rejimlerini kullanan ileri çalışmalar araştırılmaktadır. 2012 yılında yapılan bir gözden geçirme çalışması, ALT/WDL'li bireylerin 5 ve 10 yıllık sağkalım oranlarının sırasıyla %100 ve %87 olduğunu bildirmiştir.
Yeni terapiler
ALT/WDL'nin yeni tedavileri, Farklılaşmamış liposarkomun Yeni tedaviler bölümünde listelenenlerle aynıdır.
Farklılaşmamış liposarkom
Farklılaşmamış liposarkomlar, vakaların ~%10'unda mevcut bir atipik lipomatöz tümör/iyi diferansiye liposarkom (ALT/WDL) tümöründe gelişen veya bölgede bir ALT/WPL tümörünün cerrahi olarak çıkarıldığı malign tümörlerdir. Bir olan kişiler de novo tanı bu tümörün bir dediferansiye liposarkom ilerlemiştir ama böyle retroperitonda veya karın boşluğunda olarak son derece sekestre sitedeki asemptomatik geliştirilen çünkü fark edilmemeye giden bir ALT / WDL olmuş olabilir. Farklılaşmamış liposarkom tümörlerinin çoğu klinik ve genetik özellikleri ALT/WDL tümörlerinde bulunanlara benzer.
Sunum
Farklılaşmamış lipoosarkomlar (DDL) en sık orta yaşlı ve yaşlı erişkinlerde görülür ve en yüksek insidansı altıncı ila sekizinci dekattır. Nadiren, bu tümörler çocuklarda ve ergenlerde gelişmiştir. DDL tümörleri en sık retroperitoneal boşlukta ortaya çıkar, ancak ALT/WDL'ye benzer şekilde ekstremitelerde, paratestiküler bölgede, mediastende, baş veya boyunda da ortaya çıkabilir. Tüm DDL'lerin %1'den azı yüzeysel yumuşak dokularda veya göz yuvasında gelişir. Sunum sırasında, DDL tümörleri tipik olarak ağrısız, büyüktür, yıllarca yavaş ve aşamalı olarak büyümüş olabilir ve rutin X-ışınlarında kalsiyum birikimi alanları bulunur (liposarkomların Histopatolojisi bölümünde Şekil 1'de örneklenmiştir). Daha az yaygın olarak, etkilenen bireylerde tümörlerinin bir organa çarpması nedeniyle belirti ve/veya semptomlar görülür (örneğin, bağırsakların tıkanmasından kaynaklanan karın ağrısı veya üretranın tıkanmasının neden olduğu idrar yolu tıkanıklığı ). Çok nadir olarak, bir veya daha fazla belirtileri veya semptomları ile ddl mevcut olan bireyler , kronik enflamasyon (bakınız B semptomları ) ve / veya bir endokrin , nörolojik , mukokutanöz , hematolojik veya diğer doku ile ilişkili paraneoplastik sendromlar . Kronik inflamasyonun belirti ve semptomlarına ve çeşitli paraneoplastik sendromlara, tümörlerin sitokinler , hormonlar , prostaglandinler ve/veya diğer sistemik olarak etkili ajanlar salgılaması neden olur ; DDL başarıyla tedavi edildikten sonra tamamen kaybolurlar.
Patoloji
Histopatolojik ddl tümörlerin ortaya çıkması yaygın olarak değişir (liposarkomlar bölümünün Histopatoloji aşağıda. 2 bakınız Şek), ancak en sık özelliklerini sergiler farklılaşmamış pleomorfik sarkomlar (boyut yoğun değişkenliği içeren değişken ebatlı ve şekilli hücreleri ile doldurulur tümörler ve şekilli çekirdekleri ) ya da iğ hücreli sarkomlar ( bağ dokusu arka planında iğ şeklindeki hücrelerden oluşan tümörlerdir ). DDL tümörlerinin farklı kısımları genellikle arka plandaki bağ dokularının görünümlerinde farklılıklar gösterir: bu dokular miksoid olabilir (yani, standart bir H&E boyama yöntemi kullanılarak boyandığında kırmızıdan daha mavi veya mor görünen berrak, mukus benzeri bir maddeden oluşur). normal dokuların rengi) veya miksokollajen (yani miksoid arka planda yüksek kolajen lif içeriği) ve vakaların ~% 5'inde osteoid (aşağıdaki Liposarkomların Histopatolojisi bölümünde Şekil 1'e bakınız) veya kıkırdaklı materyal bulunur . Bu tümörler ayrıca hücre içeriklerinde büyük farklılıklar gösterir. Örneğin, DDL tümörlerinin %10'a kadarında ALT/WDL histopatolojisi olan alanlar bulunur ve nadir DDL vakalarında meningotelyal benzeri düz hücre kıvrımları içeren alanlar bulunur.
Genetik
Hem DDL hem de ALT/ WDL'deki neoplastik hücreler, 13 ila 15 bantlarında kromozom 12'nin q kolunun ekstra kısımlarını içeren benzer küçük fazladan markör kromozomları (sSMC'ler) ve/veya dev markör kromozomları taşır . Bu kromozomal alan, tümör gelişimi ile ilişkili iki geni içerir. , MDM2 ve CDK4 genleri. Bu iki genin fazladan kopyalarının ve/veya bunların aşırı üretilmiş protein ürünlerinin varlığı, lipomatöz tümörün başka bir tip lipomatöz tümörden ziyade bir ALT/WDL veya DDL olduğunun oldukça hassas ve spesifik bir göstergesidir. MDM2 ve CDK genlerinin ve/veya sSMC'lerde veya dev işaretleyici kromozomlardaki diğer genetik materyallerin aşırı ekspresyonunun, ALT/WDL tümörlerinin yanı sıra DDL'nin gelişimini ve/veya ilerlemesini desteklediğinden şüphelenilmektedir. ALT/WDL ve DDL neoplastik hücrelerde de aşırı eksprese edilen sMMC ve dev markör kromozomdaki diğer genler arasında HMGA2 , CPM , YEATS4 ve DDIT3 bulunur . Bununla birlikte, ALT/WDL neoplastik hücreler ile karşılaştırıldığında, DDL neoplastik hücreler: 1) iki anormal kromozomda daha yüksek gen seviyelerini ifade eder; bu, ALT/WDL'nin DDL'ye ilerlemesine katkıda bulunabilir; ve 2) bant 32'de kromozom 1'in uzun kolunda, bant 33'te kromozom 6'nın uzun kolunda ve vakaların ~%25'inde, kromozom 1'in bant 32.2'de kısa kolunda daha yüksek seviyelerde gen ürünleri bulunur. JUN geni (bu gen DDL'de aşırı eksprese edilir, ancak ALT/WDL'de değil). Yana Haziran genin ürünü, c-jun , inhibe eden hücre ölümü ve hücre çoğalmasını teşvik eden, kendi aşırı DDL ALT / WDL ilerlemesi ve / veya ddl neoplastik hücrelerin habis katkıda bulunabilir. Gen ekspresyon profili oluşturma (yani hücreler, dokular veya tümörler tarafından yapılan binlerce genin ürünlerinin ekspresyonunun ölçümü), ALT/WDL'deki adiposit hücre farklılaşması ve metabolik yolların yukarı doğru düzenlendiğini , hücre çoğalması ve DNA hasar tepki yollarının yukarı doğru düzenlendiğini ortaya çıkarmıştır. DDL'de.
Teşhis
DDL'nin histopatolojik yapısı genellikle kesin bir tanı koymak için yeterince net değildir. Bununla birlikte, DDL tanısı şu kişilerde desteklenir: tümörleri, DDL histolojik bileşenleriyle karıştırılmış ALT/WDL içeren; önceden bir ALT/WDL'ye sahip olma geçmişi olan; veya retroperitoneal liposarkom ile başvuran (DDL, tüm retroperitoneal liposarkomların ~%57'sini oluşturur). DDL tümörleri sadece nadiren (vakaların <%1'i) yüzeyel deri tümörleri olarak ortaya çıkar; göz yuvasında ALT/WDL'den neredeyse 5 kat daha az olasıdır; ve çocuklarda son derece nadirdir. Tümör hücresi MDM2 amplifikasyonunun saptanması, WDL'yi lipomlardan, displastik lipomlardan, atipik iğsi hücreli sarkomlardan, pleomorfik lipomlardan ve soliter fibröz tümörlerden ayırt etmede tanısal altın standarttır. Alternatif olarak, tümör hücrelerinde aşırı eksprese edilmiş bir CDK4 geninin tespiti veya spesifik ALT/WDL ile ilişkili sSMC'lerin veya dev markör kromozomun varlığı, DDL veya ALT/WDL tanısını güçlü bir şekilde destekler. Son iki liposarkom formu arasındaki klinik görünüm, histopatoloji ve gen farklılıkları (örneğin, cJUN geninin tümör hücresi aşırı ekspresyonu, ATL/ WDL'ye göre DDL tanısını kuvvetle destekler) genellikle aralarında ayrım yapılmasına yardımcı olur.
Tedavi ve Prognoz
Tam cerrahi rezeksiyon genellikle lokalize DDL tümörleri için önerilen birinci basamak tedavidir. Bununla birlikte, ortaya çıkan çalışmalar, bir ekstremite veya gövde ile sınırlı DDL tümörleri olan ve tahmin edilen 10 yıllık tümörle ilişkili genel sağkalım oranı %51 veya daha az olan hastaların, tedaviye kemoterapi (örn. doksorubisin artı ifosfamid ) eklendiğinde iyileştirilmiş sonuçlara sahip olduğunu göstermektedir. cerrahi rejimleri. DDL'nin bu lokalize formları için Ulusal Kapsamlı Kanser Ağı yönergelerini izleyen perioperatif radyoterapi de düşünülebilir.
Retroperitoneal DDL, DDL'nin en yaygın, cerrahi olarak değerlendirilmeyen ve ciddi formudur: tekrarlama oranı %66 ve beş yıllık genel sağkalım oranı %54'tür. Retroperitoneal DDL için birincil tedavi seçeneği cerrahi rezeksiyondur. Bir faz III klinik deney , retroperitoneal DDL tedavisinde tek başına cerrahi rezeksiyona kıyasla radyasyon tedavisinin ardından cerrahi rezeksiyonun sonuçlarında çok az fark buldu . Diğer faz III klinik çalışmalarda, erişilemeyen retroperitoneal ve/veya metastatik tümörleri olan DDL hastaları, doksorubisin ile doksorubisin artı ifosfamid veya doksorubisin ile gemsitabin artı dosetaksel ile karşılaştırılan birinci basamak kemoterapi ile tedavi edilmiştir . Diğer çalışmalar da benzer şekilde çeşitli kemoterapi rejimlerinin değerini incelemiştir. Bu çalışmalar, karşılaştırmalarında genellikle genel sağkalım sürelerinde çok az fark bulmuştur, ancak progresyonsuz sağkalım ve diğer klinik parametrelerde bazı gelişmeler göstermiştir . Bu çalışmalara dayanarak, retroperitoneal ve diğer cerrahi olarak değerlendirilemeyen veya metastatik DDL tümörleri için önerilen birinci basamak tedavi, antrasiklin bazlı kemoterapi rejimi veya tümöre dirençli veya nüksetmiş vakalarda eribulin kemoterapisidir. 2020'de yapılan bir inceleme, düşük histopatolojik dereceli ve yüksek histopatolojik dereceli DDL için medyan sağkalım sürelerinin sırasıyla 113 ay ve 48 ay olduğunu bildirdi. Tüm DDL çeşitlerinde radyoterapi, kemoterapi ve yeni tedavilerin etkinlikleri hakkında kanıt sağlamak için daha ileri çalışmalara ihtiyaç vardır.
Yeni terapiler
DDL ve daha agresif veya başka şekilde sorunlu ALT/WDL vakaları için birkaç yeni tedavi rejimi şu anda klinik deneylerden geçmektedir . Önceden tedavi görmüş veya tedavi edilmemiş DDL'li hastalarda abemaciclib'i araştıran bir faz II klinik çalışma yürütülmektedir. Ön analiz, CDK4 ve CDK6 genlerinin ürünü Serin/treonin spesifik protein kinaz enzimlerinin bu inhibitörünün , 30.4 haftalık uzun bir medyan ilerlemesiz hayatta kalma süresi ürettiğini gösterdi. Bir faz III çok merkezli, randomize, çift kör, plasebo abemaciclib klinik çalışma aktif fazda ve yakında (Temmuz 2021 belirtildiği gibi) olacak, gelişmiş tekrarlayan ve / veya metastatik DDL ile 108 bireyleri işe başlamak denetiminde olan. Çalışma, Eli Lilly ve Company ile işbirliği içinde İşbirliği Yoluyla Araştırma için Sarkom İttifakı tarafından desteklenmektedir . Ribociclib , ayrıca bir CDK4 ve CDK6 gen inhibitörü, bir mTOR inhibitörü ile kombinasyon halinde , everolimus , ileri DDL veya leiomyosarkomlu bireylerde bir faz II klinik denemesindedir . Bir faz III kayıt çalışması (yani, kesin olarak tanımlanmış bir endikasyon için düzenleyici onay almak amacıyla kabul edilebilir bir fayda/güvenlik profili oluşturmayı amaçlayan büyük bir doğrulayıcı çalışma), rezeke edilemeyen hastalarda (örn. rezeksiyonun kabul edilemez morbidite veya mortaliteye neden olduğu kabul edilir) veya en az 1 antrasiklin bazlı tedavi dahil olmak üzere önceki 1 veya daha fazla sistemik tedavide ilerlemiş metastatik DDL. Sponsor, Rain Therapeutics Inc, şu anda deneme için 160 kişiyi işe alıyor. Başka bir faz III klinik çalışma araştırmaktadır MDM2 önleyicisi milademetan karşı trabectedin , onkojenik bir engelleyici transkripsiyon faktörünün FUS-CHOP içinde, MDM2 , ALT / Wdl DDL ve -overexpressing. Milademetan, yönetilebilir toksisite ve stabil hastalık ve/veya DDL'de birkaç kısmi yanıt ile sonuçlanan bir miktar aktivite göstermiştir.
Miksoid liposarkom
Sunum
Yuvarlak hücreli liposarkom olarak adlandırılan bir liposarkom tipini içeren miksoid liposarkom (MLS), tüm liposarkomların ~%30'unu temsil eder. Çoğu çalışmada erkeklerin baskın olduğu, bireylerin dördüncü ve beşinci dekatlarında en yüksek insidansa sahiptir. Çocuklarda ve ergenlerde nadir olmakla birlikte, MLS bu yaş gruplarında teşhis edilen en yaygın liposarkom formudur. MLS tipik olarak tanıdan 1 hafta ila 15 yıl önce gelişen büyük (1 ila 39 cm; ortalama 12 cm), mobil, iyi sınırlı, ağrısız bir kitle olarak ortaya çıkar. MLS tümörleri uylukların (vakaların %65-80'i), alt bacakların (vakaların %10-15'i), retroperitonun (vakaların %8'i) ve kolların (vakaların %5'i) derin yerleşimli yumuşak dokularında bulunur. Vakaların yaklaşık üçte birinde, bu tümörler diğer yumuşak doku bölgelerine (örneğin retroperiton, toraks veya diğer ekstremite), iskelet kemiğine ve/veya akciğere metastaz yapar. Bireyler, özellikle kemikte bu metastazlarla başvurabilir; hastaların sunum sırasında X-ışınları , BT taramaları ve/veya manyetik rezonans görüntüleme dahil olmak üzere tıbbi görüntüleme yoluyla kemik metastazı açısından test edilmesi önerilmiştir .
Patoloji
MLS'nin histopatolojik analizleri (aşağıdaki Liposarkomların histopatolojisi bölümündeki Şekil 3 ve 4'e bakınız), bir miksoid matris boyunca dağılmış hücreleri (yani, bu dokular normal bağ dokusunun kırmızı renginden daha mavi veya mor görünen bir bağ dokusu arka planı) ortaya çıkarır. uygun şekilde hazırlanmış, H&E boyanmış ve mikroskobik olarak görüntülenmiştir). Bu hücreler, lipoblasts bunlardan bazıları, taşlı halka şekilli (hücre neoplastik olabileceğini düşündürmektedir bir şekli), oval şekilli veya yuvarlak şekilli. MLS tümörleri hiperselüler olabilir ve tüm hücrelerin en az %5'ini oluşturan katı yuvarlak hücre tabakalarını veya bir tavuk teli modeline benzeyen kıvrımlı kılcal damarların bir arka planında yumuşak çekirdekli ve <%5 yuvarlak hücreli hücrelerle dolu düşük hücreselliği içerebilir. En az %5 yuvarlak hücre içeren tümörler yüksek dereceli, < %5 yuvarlak hücre içeren tümörler düşük dereceli olarak sınıflandırılır. Yüksek dereceli MLS tümörleri, tipik olarak düşük dereceli MLS tümörlerinden daha agresif bir klinik seyir izler.
Genetik
MLS tümör hücreleri, vakaların >%95'inde meydana gelen bir FUS-DDIT3 füzyon geninin ( kimerik gen olarak da adlandırılır ) veya vakaların <% 5'inde meydana gelen bir EWSR1-DDIT3 füzyon geninin ekspresyonu ile neredeyse tanımlanır . FUS-DDIT3 bir sonucu olarak bir füzyon geni formları translokasyon yeri arasında: (p11) olarak adlandırılan t (00:16) (q13) DDIT3 kromozom 12'in q kolu bandı 12, gen ve yerinde FUS genindeki kromozom 16'nın kısa kolundaki bant 11 ( p arm olarak da adlandırılır ). Füzyon proteini , bu kimerik (olarak da adlandırılır şimerik bir protein) ürün onkogen gen FUS-DDIT3, yağ hücresi olgunlaşması, durdurur ve neoplasianın teşvik ettiği bilinir. EWSR1-DDIT3 füzyon geni (adlandırılır t (12; 22) (q13; q12)) 'in bir translokasyon sonuçları EWSR1 ile kromozom 22'in en q koluna bant 12.2 bulunan gen DDIT2 geni. FUS-DDIT3 füzyon proteini gibi EWSR1-DDIT3 geninin füzyon protein ürünü neoplaziyi destekler. Bu füzyon gen ilişkilerine rağmen, MLS tümörlerinin gelişimine ve/veya sürdürülmesine katkılarını tanımlamak için daha fazla çalışmaya ihtiyaç vardır.
Teşhis
Düşük dereceli ve orta dereceli MLS tümörleri, bir miksoid stroma boyunca dağılmış farklı tavuk teli damar sisteminin klasik morfolojisi ile histolojik olarak tanımlanabilir. Bununla birlikte, yüksek dereceli MLS tümörlerini diğer yuvarlak hücreli neoplazmalardan, özellikle bu klasik vasküler-miksoid paterni gizleyecek kadar yaygın hücre ve/veya saf yuvarlak hücre morfolojisinden oluşan yüksek dereceli MLS tümörlerinden ayırt etmek zor olabilir. Bir Tespit DDIT3 ile geni yeniden FUS veya EWSR1 ile genin , in situ hibridizasyon ve immünohistokimya veya RNA, bu genlerin füzyon transkriptlerinin gerçek zamanlı polimeraz zincir reaksiyonları teyit yüksek dereceli teşhisi hem de düşük dereceli çok belirsiz durumlarda ya da orta dereceli MLS tümörleri.
Tedavi ve prognoz
MLS tipik olarak cerrahi rezeksiyonla tedavi edilmiştir, ancak daha radikal müdahaleler gerektirebilir, örneğin bir uzvun nörovasküler demeti tehlikeye girdiğinde uzuv amputasyonu gerekebilir . Ameliyattan sonraki 3 yıl içinde ameliyat sonrası nüks riskinin, tüm tümör çıkarılmadığında ~%15 ve tümörün çıkarılması tamamlandığında ~%10 olduğu bildirilmiştir. Cerrahi rezeksiyona radyoterapinin eklenmesi, MLS tümörlerinin lokal kontrolünü iyileştirdi ve rezeke edilemeyen ve tekrarlayan MLS'yi tedavi etmek için önerildi. Bununla birlikte, çeşitli MLS çeşitlerinin tedavisinde radyoterapinin değerini belirlemek için daha fazla çalışmaya ihtiyaç vardır. Daunorubisin , dakarbazin ve/veya trabektedin gibi bir antrasiklin olan ifosfamid kullanan kemoterapi rejimleri faydalı bulunmuştur: bir faz III klinik deneme, trabektedin veya dakarbazin ile tedavi edilen MLS hastalarında progresyonsuz sağkalım sürelerinin sırasıyla 5.6 ve 1.5 ay olduğunu göstermiştir. 2015 yılında Gıda ve İlaç Dairesi , trabektedini, rezeke edilemeyen ve metastatik liposarkomlarda kullanım için onayladı.
Genel olarak, MLS bireylerinin 10 yıllık sağkalım oranı, diğer liposarkom formlarından kayda değer ölçüde daha uzun bir hayatta kalma oranı olan %77 olmuştur. Düşük riskli MLS ile karşılaştırıldığında, yüksek riskli MLS (tümör yuvarlak hücre içeriği ve/veya diğer olumsuz prognostik göstergeler tarafından tanımlanan risk), artan metastaz oranları ve dolayısıyla daha kısa bir hayatta kalma süresi ile ilişkilidir. Artan tümör boyutu (≥ 10 cm), daha yüksek dereceli MLS ve dolayısıyla daha kısa bir hayatta kalma süresi ile güçlü bir şekilde ilişkilidir. MLS'de olumsuz sonuçlarla ilişkilendirilen diğer faktörler arasında tümör nekrozu varlığı, >45 yaş, P53 geninin aşırı ekspresyonu ve erkek cinsiyet yer alır. Miksoid liposarkomların yuvarlak hücreli formunun da nispeten kötü bir prognoza sahip olduğu görülmektedir: çeşitli retrospektif incelemelerde, miksoid liposarkomun genellikle düşük dereceli olduğu ve bu nedenle kemoterapiye nispeten yanıt verdiği bulunurken, yüksek dereceli (yani yuvarlak hücreli} miksoid liparkom daha yüksek oranlara sahipti. metastaz, daha agresif davrandı ve kemoterapiye iyi yanıt vermedi Bununla birlikte, pediatrik hastalardaki hemen hemen tüm miksoid liposarkom vakalarının mükemmel prognoza sahip olduğunu belirtmek önemlidir.
Yeni terapiler
Bir PPAR-y agonisti (yani aktivatör), efatutazon, çeşitli ileri evre maligniteleri olan bireyler üzerinde küçük bir faz I denemesinde incelenmiştir. İlaç, MLS'li bir kişide, PPAR-y agonistlerinin bu hastalığın tedavisi için yararlı olacağını düşündüren, belirgin şekilde kalıcı bir yanıt üretti. İtalya'da yürütülen bir evre II klinik deney , stabil MLS tümörleri olan bireylerde bir trabectedin artı pioglitazonun (başka bir PPAR-y agonisti) etkilerini inceliyor . Çalışma iki ardışık adımdan oluşmaktadır. İlk adım, tek başına trabectedin ile en az 4 siklus için tedavi edilen hastaların yanıtını inceler. Stabil hastalık elde edilirse, ikinci adım, başlangıçta yanıt veren hastaların bir trabektedin ve pioglitazon kombinasyonu ile daha fazla tedavi edilmesinin etkilerini inceleyecektir. Metastatik veya rezeke edilemeyen MLS'de sirolimus (bir MTOR inhibitörü ; sirolimus ayrıca rapamisin olarak da bilinir) artı siklofosfamidin (bir kemoterapi ilacı) etkinliğini değerlendirmek için bir evre II klinik denemesi tamamlanmak üzeredir . Bir faz II klinik deneme, hastaları, yumuşak dokuların birinci basamak tedavisi olarak iki kemoterapi ilacı, doksorubisin ve ifosfamid ile kombinasyon halinde sintilimabı ( hücrelerin yüzeyinde yer alan programlanmış hücre ölümü proteinine 1 karşı yönlendirilen bir insan IgG4 monoklonal antikoru) değerlendirmek üzere toplamaktadır . MLS dahil doku sarkomları.
T hücreleri , belirli tümör türlerinde neoplastik hücrelerin yüzeyinde yer alan bir HLA-A*02 MAGE-A4 içeren peptit üzerinde eksprese edilen MAGE-A4 antijenini hedef alacak şekilde genetik olarak tasarlanmıştır . Bu tasarlanmış hücreler (ADP-A2M4-T hücreleri olarak adlandırılır) , bu antijeni taşıyan çeşitli kültürlenmiş insan kanser hücrelerine saldırdı ve öldürdü ve klinik bir aşama 1 çalışmasında, tümörleri bu antijeni eksprese eden neoplastik hücreler içeren hastalarda çeşitli katı tümör tiplerini küçülttü. Bir faz II klinik çalışma, metastatik veya ameliyat edilemez, ileri evre MSGE-4'lü HLA-A*02-pozitif hastalarda ADP-A2M4 T hücrelerinin (alıcının kendi T hücrelerinden tasarlanmış) etkinliğini ve güvenliğini araştırmak için bireyleri işe alıyor. -pozitif MLS tümörleri.
pleomorfik liposarkom
Sunum
Tüm liposarkom vakalarının %5 ila %10'unu oluşturan pleomorfik liposarkomlar (PLS), hızlı büyüyen, genellikle büyük (>5 cm) ve ağrısız fakat oldukça malign adiposit tümörleridir. Kadınlarda baskın olmak üzere, esas olarak >50 yaş bireylerde görülürler. PLS tümörleri çocuklarda nadiren bulunur. Bir bacak veya kolda (vakaların %65'i), retroperitonda veya karında (vakaların %15'i) veya nadir durumlarda gövde duvarı, spermatik kord , baş ve boyun bölgeleri, göğüs duvarı, pelvik boşluk , pulmoner plevrada bulunan PLS tümörleri , perikard ve omurga. Bu tümörler genellikle derin yumuşak dokularda lokalizedir ve vakaların sadece %25'i deri altı dokularda ortaya çıkar. Nadir PLS vakaları, etkilenen kişileri çeşitli kanser türlerini geliştirmeye yatkın hale getiren iki kalıtsal genetik bozukluk olan Li-Fraumeni veya Muir-Torre sendromlu bireylerde sunulmuştur .
Patoloji
PLS tümörlerinin histopatolojisi sıklıkla farklılaşmamış hücreler içeren alanlarla karışık miksoid liposarkoma benzeyen alanlardan oluşur. Bu tümörler hiperselüler olarak işaretlenir ve pleomorfik çekirdeklere sahip en azından bazı değişken şekilli lipoblastlar içerir. Nekroz alanları yaygındır, bazıları çok çekirdekli ve/veya yutulmuş nötrofiller içeren dev hücreler bazen mevcuttur ve bazı hücrelerde hiyalin damlacıkları görülebilir ve ayrıca tümör boyunca hücre dışı olarak dağılmış olabilir. Bu tümörlerin farklılaşmamış bileşeni çoğunlukla iğ şeklindeki hücrelerden oluşur ve vakaların %25'i epiteloid hücre morfolojisine sahip hücreleri gösterir . Bu tümörler, daha önce malign miksoid fibröz histiyositom olarak adlandırılan bir tümör olan yüksek dereceli miksofibrosarkom tipi histiyositomlara benzer bir histopatolojiye sahip en azından bazı odaklara sahiptir .
Genetik
PLS neoplastik hücreleri, çeşitli gen ve kromozom anormallikleri içerir: TP53 geni, vakaların %17-60'ında silinir veya mutasyona uğrar; RB1 gen vakaların% 60 silinir; ve Neurofibromin 1 geni, vakaların %8'inde mutasyonların inaktive edilmesiyle veya daha nadir vakalarda kromozom 12'nin uzun kolundaki bant 11.2'deki yerinin silinmesiyle kaybolur . Bu hücreler ayrıca şu bantlar 12'deki genetik materyalde kazançlar gösterebilirler. –15 kromozom 5'in kısa kolunda; kromozom 1'in kısa kolundaki bant 21; ve kromozom 7'nin uzun kolundaki bant 22. Bu anormallikler tarafından indüklenen gen kopya sayılarındaki değişiklikler , histiyositomların miksofibrosarkom tipinde görülenlere benzerdir . PLS'yi teşvik etmede gen kopya sayılarındaki bu değişikliklerin rolü/rolleri tanımlanmamıştır. Bu nedenle, PLS, neoplastik hücrelerinin, hastalığı yönlendiren karakteristik genomik değişiklikler veya tanımlanabilir genler olmadan karmaşık bir genoma sahip olması bakımından diğer liposarkomlardan farklıdır. İfadesindeki değişiklikleri algılama TP53, RB1 ve nörofibromin 1 (örneğin, PLS genlerin yanı sıra, diğer, daha az yaygın olarak değiştirilmiş genler PIK3CA , tirozin protein Syk kinazı , PTK2B , EPHA5 ve ErbB4 ), desteğe yardımcı olabilir, ancak bir tümörü açıkça PLS olarak tanımlamayın. Kromozom telomerinin uzaması, telomerlerin alternatif uzaması olarak adlandırılan patolojik mekanizmalarla sona erer , PLS vakalarının ~%80'inin neoplastik hücrelerinde meydana gelir, ancak diğer dört liposarkom formunda çok daha az yaygındır veya görülmez.
Teşhis
PLS tanısı, sunumuna, histopatolojisine ve genetiğine bağlıdır. PLS'nin histopatolojisi sıklıkla miksofibrosarkomunkine çok benzer, ancak pleomorfik lipoblast içeriği ile bu tümörden ayırt edilir.
Tedavi ve prognoz
PLS için ana tedavi radikal cerrahi rezeksiyondur; aynı zamanda organ ve dokuların sıkışmasına bağlı semptomları hafifletmek için önemli bir palyatif müdahaledir. Cerrahi, böbrek veya kolon gibi sıkıştırılmış bir organın tamamının çıkarılmasını gerektirebilir. Ancak bu ameliyattan bağımsız olarak lokal nüks oranları çok yüksektir. Radikal cerrahi ile birlikte kemoterapi ve/veya radyoterapi kullanımının sağkalımı uzattığı gösterilmemiştir ve tartışmalı müdahaleler olarak kabul edilmektedir. Ulusal Kapsamlı Kanser Ağı radyasyon terapisi ile kombine uygulanabilir, tam cerrahi rezeksiyon ile yüksek riskli lokalize PLS bireyler için tedavi önerir. Metastatik hastalığı olan bireyler, farklılaşmış liposarkom için kullanılan rejimlere benzer kemoterapi (örn. doksorubisin artı ifosfamid veya eribulin ) ile tedavi edilmiştir (bu liposarkom tipinin tedavisi ile ilgili yukarıdaki bölüme bakınız) PLS tümörlerinin yaklaşık %20'si uzak bölgelere metastaz yapar, çoğu yaygın olanları akciğer (metastazların %82'si), karaciğer (metastazların %18'i) ve kemik veya pankreastır (metastazların %18'i). 1, 3 ve 5 yıllık PLS sağkalım oranları sırasıyla %93, %75 ve %29 olarak bildirilmektedir. Gövdenin ortasında yer alan, 10 cm'den büyük, derin yerleşimli veya nekroz alanları içeren tümörler daha kötü prognoza sahiptir.
Miksoid pleomorfik liposarkom
Miksoid pleomorfik liposarkom (başlangıçta pleomorfik miksoid liposarkom olarak adlandırılır) ilk olarak 2009'da liposarkomlarla ilgili büyük bir çalışmada tanımlanmıştır. Başlangıçta pleomorfik özelliklere sahip miksoid liposarkomların olası bir varyantı olarak kabul edilirken , Dünya Sağlık Örgütü (2020) onu liposarkomların yeni ve farklı bir formu olarak sınıflandırdı. Bu sınıflandırma, miksoid pleomorfik liposarkomların, miksoid liposarkomlara benzer histopatolojik özelliklere sahip olmakla birlikte, diğer üç liposarkom formundan olduğu gibi miksoidden de farklı klinik ve en önemlisi kritik genetik ve moleküler özelliklere sahip olduğu bulgularına dayanmaktadır.
Sunum
Miksoid pleomorfik liposarkom (MPL), çocuklarda, ergenlerde, genç erişkinlerde ve daha yakın tarihli bir çalışmada 50 yaşın üzerindeki bireylerde gelişen liposarkomların son derece nadir ve oldukça agresif bir şeklidir. MPL tümörleri sıklıkla mediastende ve daha az sıklıkla ekstremitelerde, baş ve boyunda, karın boşluğunda veya gövdede bulunan derin yumuşak doku kitleleri olarak ortaya çıkar. En az iki MPL vakası, bireyleri çeşitli kanserler geliştirmeye yatkın hale getiren kalıtsal bir genetik bozukluk olan Li-Fraumeni sendromu ile bireysel olarak sunulmuştur .
Patoloji
Histopatolojik analizlerde MPL tümörleri, konvansiyonel miksoid liposarkoma benzeyen alanlardan oluşur; Toplam tümör alanlarının %30-50'sini temsil eden bu alanlar, bol miktarda miksoid matriks, iyi gelişmiş bir kapiller damar sistemi, yuvarlak ve/veya hafif iğ şeklinde yumuşak hücreler, vakuollü lipoblastlar ve küçük şekilli çok çekirdekli hücrelere sahiptir. Çiçekler. Bununla birlikte, bu alanlar aynı zamanda, hücrelerde görülen miksoid liposarkom tümörlerinden daha yüksek derecelerde nükleer genişleme ve düzensizlik gösteren oldukça pleomorfik hücrelerin bir saçılımını da içerir. MPL tümörlerinin diğer alanları daha hücreseldir ve hızla büyüyen ve yüksek oranda pleomorfik lipoblastlardan oluşur .
Genetik
MPL'deki neoplastik hücreler, miksoid fibrosarkom vakalarının sırasıyla >%95 veya <% 5'inde neoplastik hücreler tarafından ifade edilen FUS-DDIT3 veya EWSR1-DDIT3 füzyon genlerini ifade etmez . RB1 tümör baskılayıcı geninin delesyonu veya patolojik baskılanması nedeniyle inaktivasyonu tüm vakalarda MPL bulunur. MPL neoplastik hücreler ayrıca kromozomlarında yaygın olarak başka değişikliklere de sahiptir. Normalde 1, 6, 7, 8, 19, 21 ve/veya X kromozomlarında bulunan bazı genetik materyalde anormal kazançlar ve normalde 2, 3, 4, 5, 10 kromozomlarında bulunan genetik materyalde kayıplar gösterebilirler. , 11, 12, 13, 14, 15, 16, 17 ve / veya 22 13 kromozomunun uzun kolu üzerinde bant 14 kayıp genetik materyali içeren RP1 gen değil, aynı zamanda RCBTB2 , DLEU1 , ve ITM2B genleri. Nadir olması ve daha yeni tanımı nedeniyle, bu genetik anormalliklerin moleküler özellikleri ve önemi henüz tam olarak tanımlanmamıştır. Bununla birlikte, çalışmalar, RB1, RCBTB2, DLEU1 ve ITM2B genlerinin herhangi birindeki veya daha fazlasındaki , ancak özellikle RP1 genindeki Kayıpların, MPL'nin gelişimine ve/veya ilerlemesine katkıda bulunabileceğini göstermiştir.
Teşhis
MPL tanısı, tümörlerinin klinik görünümüne, miksoid liposarkoma histopatolojik benzerliğine ve en kritik olarak neoplastik hücrelerinde FUS-DDIT3 sn EWSR1-DDIT3 füzyon genlerinin bulunmamasına bağlıdır .
Tedavi ve prognoz
MPL'li bireyler tümörlerini çıkarmak için cerrahi rezeksiyonla tedavi edilirken, 2021'de yapılan bir inceleme, MPL için radyasyon ve kemoterapi rejimlerine (tek başına veya ameliyatla birlikte kullanıldığında) ilişkin standart bakım önerilerinin bulunmadığını buldu. Bu tümörleri tedavi etmek.
Liposarkomların histopatolojisi
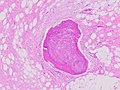
Şekil 1 Bir liposarkom tümöründe kemik oluşumunun mikrografı

Şekil 2 Farklılaşmamış bir liposarkom tümörünün mikrografı

Şekil 3 Miksoid liposarkom tümörünün düşük güçlü mikrografı

Şekil 4 Miksoid liposarkom tümörünün yüksek güçlü mikrografı


.jpg)
.JPG)
Tıbbi Görüntüleme
Liposarkomların tıbbi ultrasonografisi ve manyetik rezonans görüntülemesi (MRI), bunların kapsamını, cerrahi erişilebilirliğini ve gözlemlenen herhangi bir organ işlev bozukluğuyla ilişkisini belirlemede yardımcıdır ve sıklıkla gereklidir. Ultrasonografi genellikle bir liposarkomu iyi huylu bir lipomdan ayırt edemediğinden, MRG, bu ayrımı yapmakla ilgili kanıtlanmış kanıtlar için tercih edilen ilk görüntülemedir.

Şekil 5 Lipomatöz matriksinden yansıyan yüksek eko alanları ve lipomatöz olmayan alanlarından yansıyan düşük eko alanları olan bir liposarkomun ultrasonografisi .
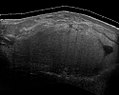
Şekil 6 Lipomu taklit eden bir liposarkomun ultrasonografisi . Bu homojen yüksek ekoik kütle, bir lipom ile aynı görünüme sahiptir.

Şekil 7 Tümörün bu yatay kesitinde, 40 yaşındaki bir erkeğin sol aksiller bölgesindeki yüksek dereceli miksoid liposarkomun MRG'si , beyaz rengiyle vurgulanmıştır.



Toplum ve kültür
Önemli vakalar
- Chad Brown (1961-2014), bir poker oyuncusu, liposarkomdan öldü
- Teorik fizikçi Richard Feynman (1918–1988), hastalığı tedavi etmek için geçirdiği ameliyatın ardından öldü.
- Rob Ford (1969-2016), eski Toronto belediye başkanı ve Toronto belediye meclisi üyesi, pleomorfik liposarkomdan öldü.
- Hokie Gajan (1959-2016), eski New Orleans Saints için koşan ve takım için radyo renk yorumcusu, liposarkomdan öldü.
- Charlie Davies (1986-), Philadelphia Major League Soccer Birliği'nin eski futbolcusu , 2016'da liposarkom teşhisi kondu.
- Mark Strand (1934-2014), eski ABD Şair Ödülü sahibi ve Pulitzer Ödülü sahibi, liposarkomdan öldü.
Ayrıca bakınız
- lipom
- Wendy Walk , misyonu liposarkom da dahil olmak üzere sarkomlar için fon ve farkındalık yaratmak olan kar amacı gütmeyen bir kuruluş
Referanslar
Dış bağlantılar
| sınıflandırma | |
|---|---|
| Dış kaynaklar |